The Spark Program will not be accepting applications for the 2025–2026 cycle, as the program will be on pause during that time. We encourage you to stay tuned for future opportunities and related open calls!
CDI Spark Award
Award Information
The goal of the CDI Spark Award is to support basic science research trainees in their projects by helping them leverage data science and machine learning expertise to uncover previously unseen trends in their data.
The CDI Spark Award will provide trainees with the expertise they need to conduct innovative research and create essential links within the scientific community, to continue to make impactful discoveries throughout their careers, and help fuel leaders of Canada’s scientific community.
The award aims to support basic research trainees across the UHN community by:
- Providing funding and resources to support their research projects
- Providing expertise and mentorship in data science and machine learning to better answer their research questions
- Fostering connections across research teams to provide guidance, build collaborations, and create a community
The CDI Spark Award will support four (4) trainees for the 2024-2025 academic year. Each successful trainee will receive $20,000 towards their yearly salary
FREQUENTLY ASKED QUESTIONS
Program Support
The program consists of the following components:
- Salary funding – $20K per participant
- Access to CDI specialists
- Educational webinars
- Research day
Expectations
As part of the Spark Award, trainees will be expected to contribute outputs of their research project to the CDI Spark Registry. Any open-source code, data, publications or conference presentations from the Spark-funded project will be shared with the CDI Spark team to add to the internal Spark Registry.
There will also be a commitment from the trainee’s PIs as part of the Spark Award. PIs are expected to:
- Attend the Spark Research Day and provide a 5-10m introduction to their lab’s research before their trainee presents their project
- Become a part of the adjunct specialist resource, wherein PIs are available to meet with Spark trainees from outside their labs if the trainees have questions relevant to their discipline (commitment: 30m, if requested by a trainee)
Funding
The program will support trainees with $20,000 in salary funding provided in two instalments, first at program kick-off and second at the program midpoint.
CDI Specialists
Trainees will have access to CDI specialists for group and individual training sessions. CDI has specialists across a variety of domains:
- Machine learning
- Computer science
- Data science
- Data governance and accessibility
- Navigating UHN Digital
- Service design
- Project management (waterfall/PMP, Lean Six Sigma, Agile)
Educational Webinars
There will be three webinars throughout the program, presented by CDI specialists. Trainees will provide topic suggestions from specialist domains of expertise at the start of the program. The series will be tailored to their learning goals. The webinars are open to the lab of the trainees.
Research Day
As part of project close-out, trainees will give a 15 minute presentation to discuss their project outcomes and lessons learned. These presentations will celebrate trainees’ skill development and accomplishments during the program. This session will be open to the labs of trainees as well as the CDI team.
CDI Spark Award Information Session
Recording of our information session for the 2022 application cycle.
Eligibility Criteria
Candidate
- The award will target 2nd and 3rd year Ph.D. students or post-doctoral fellows in a lab of a Scientist or Clinician-scientist with an appointment at UHN or PM.
- Experience with computational data manipulation. Preference for people with formalized coding experience (i.e., R, Python, MatLab, etc)
- Domestic and international students are eligible to apply
- For more information, please refer to the FAQ linked below
Project
- Research projects must be focused on pre-clinical or basic research
- Projects must be in the field of cancer research
Important Dates
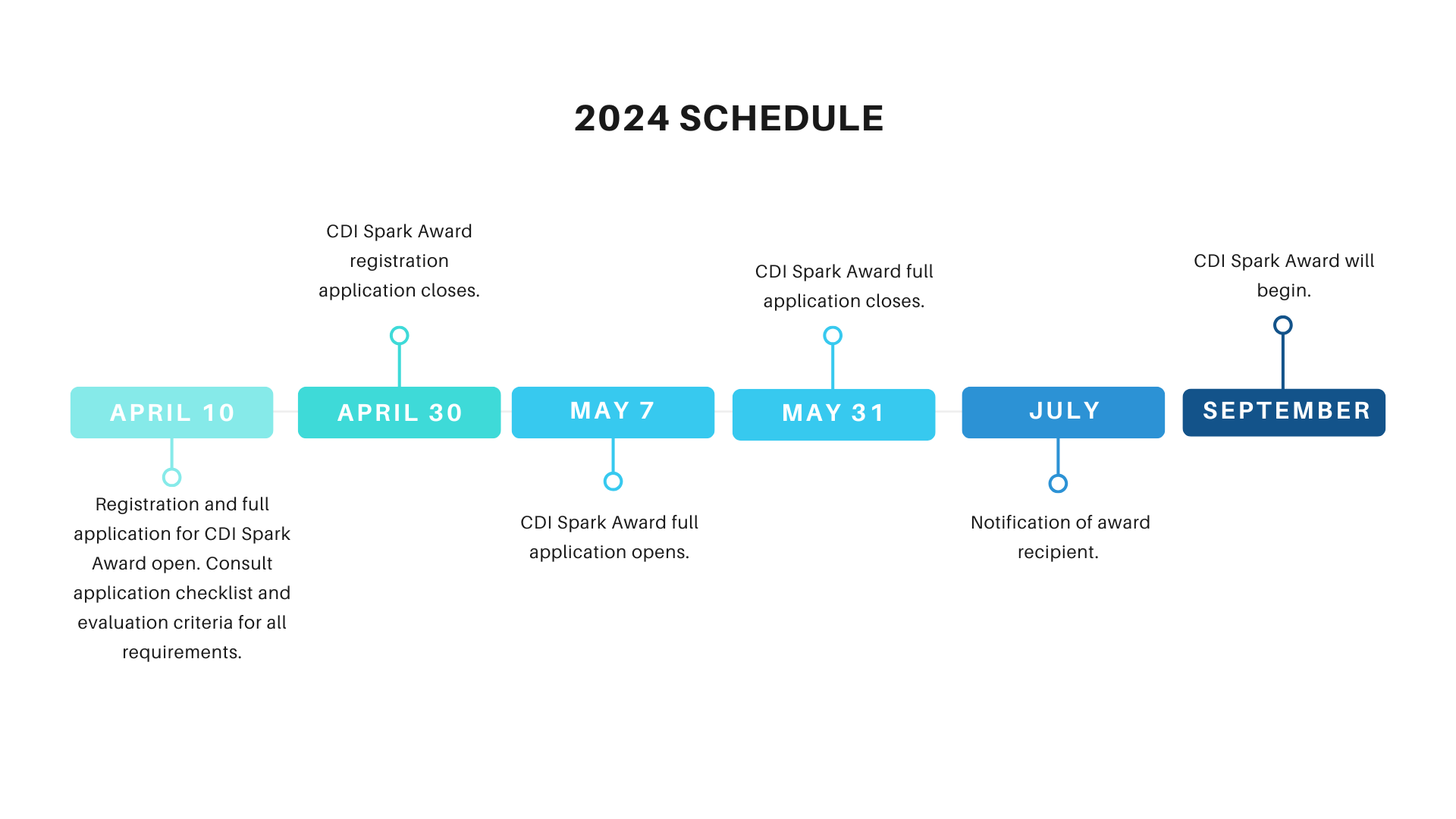
How to Apply
Follow the notes below for your application. Please submit all documents via email in PDF format.
REGISTRATION APPLICATION
- Use the email subject “CDI Spark Award Registration [Last name, First name]” to submit the registration form
- Registration for the 2024-2025 cohort is now open!
REGISTRATION FILLABLE PDF
FULL APPLICATION
- Use the email subject “ CDI Spark Award Full Application [Last name, First name]” to submit the full application form
- Applications for the 2024-2025 cohort are now open!
FULL APPLICATION
APPLICANT CHECKLIST
EVALUATION CRITERIA
2024 Cohort
 Mohamed Alias
Mohamed Alias
Mohamed is a 2nd year PhD student in Dr. Robert Kridel’s laboratory within the Department of Medical Biophysics at the University of Toronto. His doctoral research focuses on investigating cell-free DNA as a noninvasive biomarker for cancer detection and classification in lymphoma. Through liquid biopsy, we can investigate the characteristics of cell-free DNA in blood plasma such as methylation and utilize it as a screening tool for the noninvasive detection of cancer. We are also able to utilize cell-free DNA methylation to classify our lymphoma samples into it’s specific subtype thereby paving the way for precision cancer medicine. Mohamed hopes to translate novel discoveries from the lab into useful tools for clinical application. Outside the laboratory, Mohamed enjoys reading and outdoor activities such as hiking.
Lay abstract:
Lymphoma is a cancer of white blood cells and is classically categorized into non-Hodgkin and Hodgkin lymphoma. Depending on the stage and subtype involved treatments for it range from chemotherapy, radiation therapy, immunotherapy, and surgery. It is typically diagnosed by obtaining a tissue sample from the lymph nodes, which is invasive. Novel noninvasive methods would include obtaining a patient’s blood sample and looking at cell-free DNA released from cancer cells. My project will investigate cell-free DNA methylation for the noninvasive detection of lymphoma. These cell-free DNA released from cancer cells into the bloodstream can be detected through assays such as cell-free methylated DNA immunoprecipitation sequencing (cfMeDIP-seq). I will utilize the unique methylation signature of cell-free DNA of lymphoma cells to differentiate it from the cell-free DNA of healthy cells for detection. Since the methylation signatures are remarkably different between cancer and healthy cells, I will utilize their distinctive signature as a molecular fingerprint to classify the lymphoma cases into their respective subtypes using machine learning approaches. My work aims to aid physicians in diagnosing lymphoma noninvasively in patients from the clinic.
 Fatemeh Zabihollahy
Fatemeh Zabihollahy
Fatemeh is an AI research scientist at the Toronto General Hospital Research Institute, working under the guidance of Dr. George Yousef and Dr. Neil Fleshner. Her research focused on the development of multi-modal AI for cancer diagnosis and prognosis. She graduated from Carleton University, Ottawa, Canada, in April 2020, where her Ph.D. work entitled “Deep Learning Methods for Abnormality Detection and Segmentation in Computed Tomography and Magnetic Resonance Images (MRI)” has been awarded the University Medal for outstanding graduate work at the doctoral level. Fatemeh then joined Johns Hopkins University as a postdoctoral scholar to design and implement deep learning-based methods for real-time MRI-guided robotic surgery systems for abdominal and body intervention. She moved to the University of California, Los Angeles as a project scientist to study and develop AI-based techniques for multi-modality image registration.
Lay Abstract
Lymph node metastasis (LNM) is associated with a higher risk of prostate cancer (PCa)-related death, and poor prognosis, and requires modification of the treatment plan. Manual assessment of LNs on glass slides is a time-consuming task that significantly increases the workload of pathologists. It is also prone to error as the human eye might not be able to identify very small metastatic deposits. The recent transition to digital pathology, combined with breakthroughs in deep learning (DL), has the potential to help pathologists accurately examine LNs through whole-slide images (WSIs). Training DL-based methods, however, require a large number of labeled data. My research goal is to adopt a contrastive-based few-shot learning (CFSL) framework to develop an AI tool for autonomous analysis of LNM using limited labeled WSIs. I previously used FSL for breast cancer diagnosis using a handful of manually annotated MRIs and demonstrated that the FSL-based method significantly outperforms radiologists for differentiating benign and malignant tumors (p<0.001). I hypothesize that CFSL can be adopted for accurate and fast analysis of LN WSIs that obviates the need for mass data labeling. The outcome of this research might provide an improved diagnosis for patients with PCa.
 Zhongyuan (Jasper) Zhang
Zhongyuan (Jasper) Zhang
Zhongyuan (Jasper) Zhang is a second-year Ph.D. student in Dr. Wei Xu’s lab at the Dalla Lana School of Public Health, University of Toronto. He obtained his Master of Science in Biostatistics from the University of Toronto and an Honours Bachelor of Mathematics from the University of Waterloo. Jasper’s doctoral research focuses on integrating multimodal data, including medical imaging, genomics, etc., to study multiple outcomes. He has research experience in Survival Analysis, Deep Learning, and Health Economics. In his spare time, Jasper is a tenor in the UofT Allegro Choir. He enjoys playing soccer and badminton, cooking fusion cuisines, and reading interesting papers.
Lay abstract:
Cancer is a complex disease that varies widely from patient to patient, making personalized treatment crucial. However, researchers often lack large, detailed datasets that reflect this diversity. Our project addresses this by using advanced machine learning to create synthetic data that mimics real cancer patients’ multi-omics data (such as gene expression and copy number variations). We use generative models, a technology that generates new data while retaining the essential characteristics of original datasets. Using The Cancer Genome Atlas (TCGA) as a foundation, we produce synthetic datasets that accurately represent cancer progression and treatment responses. This approach broadens our ability to study cancer across diverse patient groups without new, costly experiments and protects patient privacy. By addressing critical data shortages, our project offers a scalable, ethical solution that could significantly advance personalized medicine, enhancing our understanding and treatment of cancer.
2023 Cohort
 Mary Agopian
Mary Agopian
Mary is a second-year doctoral candidate in Dr. Michael Hoffman’s laboratory within the Department of Medical Biophysics at the University of Toronto. Her doctoral thesis focuses on improving current methods of characterizing and quantifying T cell receptor diversity. By understanding and quantifying the process of T-cell receptor generation, she aims to develop a diagnostic test for T-cell lymphomas that is effective, rapid, and non-invasive. Mary graduated with an Honours Bachelor of Science degree in Microbiology and Immunology from McGill University. Her background in immunology, combined with her academic interest in computational medicine and bioinformatics, allows her to conduct this interdisciplinary research at the intersection of immunology, genetics and bioinformatics. Outside of work, you can find Mary hiking and identifying mushrooms for the citizen science effort of documenting fungal species in North America.
Lay abstract:
Clinicians often use bone marrow aspiration and biopsy to diagnose lymphomas. However, T-cell lymphomas are less likely to spread to the bone marrow, meaning this diagnostic method can lead to incorrect test results, delays in treatment, and additional invasive testing for patients. For instance, it can take up to 5-6 years for physicians to diagnose cutaneous T-cell lymphoma from the first symptoms.
One of the methods clinicians use to diagnose a patient’s specific type of lymphoma is T-cell receptor repertoire sequencing (TCR-seq). TCR-seq is used to investigate T-cell genes for abnormalities associated with specific lymphoma subtypes. TCR-seq can detect lymphoma-specific patterns, making it a potential tool for diagnosing T-cell lymphoma. However, measuring and comparing TCR-seq results is challenging, which limits their usefulness for diagnostic tests. But, TCR-seq’s diagnostic ability can be improved by creating controls that serve as a reference point, enabling quantitative measurements and comparisons of results. Therefore, this project aims to develop an effective, rapid, and non-invasive T-cell lymphoma diagnostic test through the creation of reference controls.

Rajesh Detroja
Rajesh is a Postdoctoral researcher at the Princess Margaret Cancer Center in Toronto, working under the guidance of Dr. Robert Kridel. He completed his Ph.D. at Bar-Ilan University in Israel, specializing in Bioinformatics. Rajesh is passionate about applying cutting-edge computational technologies to advance the scientific understanding of Cancer Biology to achieve precision medicine. During his Ph.D., he developed an efficient computational method to study expressed fusion genes and chimeric RNAs in complex diseases like Cancer and Rheumatoid Arthritis. Currently, Rajesh is using machine-learning techniques to analyze the methylation profiles of follicular lymphoma (FL) patients. This research aims to identify distinct epigenetic subtypes, which will be crucial in developing targeted treatments for FL patients. Besides research, Rajesh enjoys exercising and taking walks in nature.
Lay Abstract
Follicular lymphoma (FL) is a type of B-cell cancer that tends to relapse after treatment, putting some patients at an increased risk of early death. The current classification used for FL patients is insufficient in explaining the biological and clinical differences among them, making it challenging to develop precision medicine approaches. Thus, my research aims to identify new molecular subtypes of FL by investigating inter-patient molecular differences. As methylation alterations play a crucial role in defining cellular identities and phenotypes, I hypothesize that subgrouping patients based on genome-wide methylation profiling of FL tumour cells can distinguish FL subtypes. My specific goal is to build computational models that can effectively identify new epigenetic subtypes of FL and subsequently gain insights into the cellular origin, pathogenic mechanisms, and clinical behaviour of each subtype. Preliminary results indicate that the methylation profiles of FL samples are significantly different. For instance, I discovered that advanced cases of FL are similar to fast-growing diffuse large B cell lymphoma (DLBCL) and normal memory B-cells, while limited cases of FL are more similar to normal germinal center B-cells. Altogether, the outcome of this research will represent a critical step toward developing subtype-specific therapies for FL patients.
 Shawn Hercules
Shawn Hercules
Dr. Shawn Hercules is a highly motivated and innovative researcher with expertise in public health and molecular cancer biology. With a PhD from McMaster University focused on investigating the epidemiology and genomics of triple negative breast cancer in Nigeria, Barbados and Jamaica, Shawn is an interdisciplinary scientist passionate about advancing health equity and cultivating diversity in science. Currently, Shawn’s research at the Princess Margaret Cancer Centre is focused on identifying molecular changes in precancerous breast tissues of high-risk populations to enable early detection and prevention of breast cancer. Shawn has a track record of successful collaborations with pathologists, surgical oncologists, and bioinformaticians, and they are committed to translating their research findings into real-world solutions. Shawn is also dedicated to promoting diversity and inclusion in STEM through various outreach initiatives such as Science is a Drag.
Lay abstract:
With advances in genetic testing, females identified at high-risk for breast cancer continue to grow with limited treatment and prevention options outside of surgery. As tremendous strides have been made with capacity to detect breast cancer, a higher frequency of pre-cancerous breast masses is being detected. Though majority of cases diagnosed with some of these masses may not progress to breast cancer, most of them are treated with surgery, radiation therapy or chemotherapy. I aim to reduce overtreatment and differentiate risk between individuals at high-risk for this disease. I propose to study high-risk populations with recently developed cutting-edge technologies. I will construct a unique database with >3000 high-risk patients to investigate their clinical trajectory and identify breast tissues. I will create a high-risk breast cancer tissue resource to reveal biological factors that influence precancerous breast masses to progress. I will also expand knowledge on high-risk populations to be more inclusive of understudied high-risk populations and will build on knowledge garnered from tissue resources to develop precision medicine for prevention. With this work, I aim to uncover critical biological pathways that offer the potential to stop breast cancer before it starts.
 Vivian Chu
Vivian Chu
Vivian is a second-year PhD Student in the laboratories of Dr. Bo Wang and Dr. Hansen He in the Department of Medical Biophysics at the University of Toronto. Vivian obtained her Bachelors of Science at the University of Waterloo, specializing in Molecular Genetics and Bioinformatics. Vivian’s doctoral research leverages deep learning to enhance the design and functionality of RNA-based therapies, such as RNA interference molecules and neoantigens, to guide personalized cancer treatment strategies and understand how different factors impact gene regulation. Vivian strives to bridge the gap between complex omic data sets, which include genomics, transcriptomics, proteomics, and more, with the aim of elucidating a more comprehensive picture of biological systems. She believes that effective integration and interpretation of multi-omic data can unlock novel insights into disease mechanisms and potential therapeutic interventions. Outside of the lab, Vivian enjoys photography, camping and reading.
Lay Abstract
In diseases like cancer, sometimes genes that shouldn’t be active get switched on, or genes that should be working get turned off. One way to control these switches is using small interfering RNAs (siRNAs). These are molecules, similar to DNA, which can dictate which genes can turn into proteins. They do this by binding to messenger RNA (mRNA), which carry instructions from DNA to the machinery that makes proteins in the cell. When an siRNA binds to an mRNA, it effectively silences that gene, tagging it for degradation and stopping the production line for a specific protein. However, the design of these siRNAs difficult – their complex 3D shapes and the way the interact with other molecules can greatly affect their performance. To tackle these challenges, I am using deep learning, a powerful type of artificial intelligence that is adept at recognizing patterns and making predictions, to understand how the 3D shapes of siRNAs impact their ability to turn off genes and improve efficiency. On a basic level, this research aims to enhance our understanding of the complex interplay between RNA secondary structure and siRNA efficacy. On a translational level, this work will enable us to harness the full therapeutic potential of RNA-based therapies, for improved personalized cancer treatment.
2022 Cohort
 Emma Bell
Emma Bell
Dr. Emma Bell is a postdoctoral research fellow at the Princess Margaret Cancer Centre in Toronto, Canada. As a bioinformatician, they combine biology, statistics, and information engineering, to ask biologically and clinically meaningful questions of genomics data. Their research uses cell-free methylated DNA immunoprecipitation sequencing (cfMeDIP-seq) to detect evidence of cancer in blood plasma donated by ovarian cancer patients. This project aims to test the potential of cfMeDIP-seq as a liquid biopsy for this disease. Dr. Bell’s long-term career goal is to improve gynecologic healthcare outcomes for women, non-binary and trans people.
Abstract:
Most ovarian cancer patients receive their diagnosis when their cancer is already advanced. Only 30% of these patients survive beyond 5-years. In contrast, 92% of patients diagnosed with early-stage disease survive beyond 5-years. Early diagnosis would immediately improve the long-term survival of ovarian cancer patients. Thus, we must develop sensitive and specific diagnostic tools.
Ovarian cancer manifests as subtypes with distinct molecular drivers and differing prognoses. An effective diagnostic tool must distinguish these subtypes.
Our lab developed cell-free methylated DNA immunoprecipitation sequencing (cfMeDIP-seq) to detect early-stage cancer. Cells methylate their DNA to control its expression. Each cell type does so following a unique pattern. Cancer cells highly methylate their DNA. This allows us to enrich for circulating tumour DNA (ctDNA) from the larger pool of cell-free DNA (cfDNA). Previous cfMeDIP-seq studies could discriminate not only between patients and disease-free controls, but also closely related cell types and early- and late-stage disease.
This project will evaluate cfMeDIP-seq as a potential diagnostic tool for ovarian cancer. I hypothesise ctDNA released from an ovarian tumour reflects the DNA methylation profile of its histological subtype. I will test this using the cfMeDIP-seq method to profile blood plasma donated by ovarian cancer patients.
 Tina Keshavarzian
Tina Keshavarzian
Tina is a second-year PhD candidate at the University of Toronto, advised by Dr. Mathieu Lupien. Tina obtained her bachelor’s degree at the University of Toronto, specializing in Neuroscience with minors in Computer Science and Physiology. She loves using computers to understand biology. Her undergraduate thesis modelled neurodevelopmental trajectories in mice with autism-like behaviour. Currently, her doctoral thesis focuses on how DNA is used differently to make some cancers more aggressive than others. She attempts to integrate different layers of genetic information together to gain a more holistic understanding of cancer biology, which helps us develop better diagnostic tools and treatments. Besides research, Tina loves exercising and the outdoors. She’s part of the synchronized swimming team, trains for endurance sports, and loves backpacking and camping in the mountains.
Abstract:
The human body contains 37 trillion cells, each performing a very specific function. To do this, each cell carries out instructions from its genetic material stored as a 3 billion basepair long string of DNA. The DNA sequence is virtually identical in every cell, but the instructions carried out by each cell vastly differ. This is because different parts of the DNA are used to carry out these functions. Cancers can arise when normal cells lose control over which parts of the DNA are used. This can give rise to fast-growing cancer cells and potentially to cells that can migrate to other tissues and form metastatic tumours. Because metastasis is the reason for almost all cancer-related deaths, I intend to understand the parts of the DNA that are specifically used in metastatic cells to support their aggressive and invasive behaviour. Understanding the specific state of the 3-billion basepair DNA string in every single primary and metastatic cancer cells requires significant computational power and powerful statistical methods. Using technological advances in sequencing, we can start to pinpoint regions of the DNA that are used in metastasis, which will map the genetic basis of this deadly disease and open new avenues for diagnosis and intervention.
 Sasha Main
Sasha Main
Sasha completed her undergraduate degree at Queen’s University, where she studied Biology with a Specialization in Mathematics. She is now a second-year PhD candidate in the laboratory of Dr. Scott Bratman within the department of Medical Biophysics (MBP) at the University of Toronto. Sasha’s doctoral research focuses on fragmentation characteristics of cell-free DNA in the bloodstream, and through liquid biopsy, using these features to reveal breast cancer biology and predict treatment response to targeted therapies. Sasha hopes to translate novel discoveries from the lab into clinically useful tools for the management of cancer patients. Outside of her research, Sasha enjoys traveling and spending time outdoors, namely hiking, camping, skiing, and cycling.
Abstract:
In our bodies, cells die and release fragments of their DNA into the bloodstream. In cancer patients, DNA fragments are shed from both normal healthy cells and tumour cells into the blood circulation. By testing a simple blood draw, we can use this circulating DNA to reveal tumour-specific information throughout a patient’s journey with cancer. We aim to advance existing blood test technologies by investigating characteristics related to how the DNA was fragmented to reveal more information about breast cancer, such as its location and behaviour. For example, I will explore the lengths of these DNA fragments, the types of DNA sequence at the fragments’ ends, and the abundance of these fragments across the human genome. These features are different between cancer patients and healthy individuals, and in cancer patients can provide helpful hints about a tumour’s biology. Specifically, we will explore whether these different DNA characteristics distinguish the breast cancer subtypes. Altogether, this research will uncover new tumour-specific information in circulating DNA that can one day be used to inform cancer management, ultimately improving the lives of cancer patients.
 Derek Wong
Derek Wong
I joined the lab of Dr. Trevor Pugh in February of 2021 as a Post-doctoral fellow. My project is part of the Canada-wide CHARM consortium and involves investigating and developing blood-based cancer classifiers for the early detection of cancer in patients with hereditary cancer syndromes. Prior to this, I completed my PhD at the BC Cancer Research Center in Vancouver, BC with Dr. Stephen Yip where I investigated the proteogenomic landscape of low-grade gliomas. I am passionate about translational genomic research that can improve and impact patients using data. Outside of the lab, I enjoy rock climbing and visiting craft breweries.
Abstract
As cells in your body grow and divide, they release DNA fragments into the blood. These DNA fragments are often non-random and provide clues to the type of cell that is releasing them (ie blood cell versus liver cell). Based on this, scientists are able to analyze the patterns of DNA fragments in the blood to determine where the DNA is coming from. In the case of cancer, each type of cancer (ie breast, colon etc) releases a unique pattern of DNA that is different than healthy cells and can be used to detect if and where the cancer is in a patient. This project is focused on using machine learning to properly and effectively classify these unique patterns of DNA fragments in order to predict if and what kind of cancer a patient may have. This tool will be especially helpful for detecting early cancer (when and where) in patients with hereditary cancer syndromes who are at a high-risk of developing cancer.
Contact Us
If you have any questions about the CDI Spark Award, you can email us here: pmcdi@uhn.ca